")
")










Our specific expertise in Prion, Alzheimer‘s diseases (AD) and stem cells was the basis for our research focusing on physiological mechanisms and therapeutic approaches of these diseases as follows:
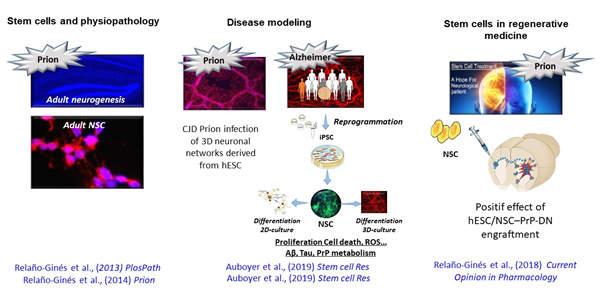
* To develop a regenerative medicine approach, we used neural stem cells derived (NSC) from embryonic stem cells (ES) that express prion protein (PrP) dominant-negative proteins (PrP-DN) to inhibit prion replication. In preclinical studies using prion infected mice we showed a decrease in the vacuolisation, astrogliosis and prion accumulation. This motivates the future transfer the technology developed in mouse ES to human ES. Our future objective is to transfer this therapeutic approach to human models with the perspective to go to clinical trials. We will use transgenic mice infected with human CJD prion and grafted with humanNSC expressing PrP-DN.
* We investigated the role of adult neurogenesis and NSC in prion propagation. We showed that endogenous NSC were not only infected by prion but also that their neuronal differentiation potential was also altered. We also showed more proliferation and apoptosis with the infected NSC. Our objective aim now at deciphering the molecular mechanism underlying these phenotype.
* While relevant cellular model for AD are crucially lacking, we generated of induced pluripotent stem cells (iPSC) by reprogramming fibroblasts from genetic AD patients linked to mutation in APP and PS1 genes. These cells cultured in 3D organoid culture systems showed typical AD Tau hyperphosphorylation and Aβ aggregates. Using the AD-iPSC, we observed some alterations as soon as the NSC developmental cellular stage (glucose uptake, cell survival, proliferation, neural differentiation, decrease of the mitochondrial biomass…). Cumulative observations based on brain onthogeny, AD clinical and research reports lead to a new etiological hypothesis that some AD lesions appearing through aging could originate from slight dysregulations during early brain development. To address this challenging hypothesis, we will use AD-iPSC, 3D neural networks and mini-brain mimicking corticogenesis