")
")










PROJECTS:
-
Identification of new genes for inherited retinal dystrophies and hereditary optic neuropathies
The identification of novel genes, in inherited retinal dystrophies and optic neuropathies, is of major concern for the pathophysiological understanding and diagnosis of these diseases, to establish phenotype/genotype correlations, for a precise diagnosis and for the improvement of genetic counseling. It is also an indispensable step in the development of innovative therapies and in particular gene therapy.
Although many responsible genes have been discovered in these diseases, about 30 to 40% of patients do not have mutations in known genes. The goal of our studies, using candidate gene strategies, genetic mapping and exome and genome sequencing, is to discover the missing genes. Thus, we will have a more complete representation of the genetics and physiopathology of these diseases, these data being necessary to produce new effective therapies.
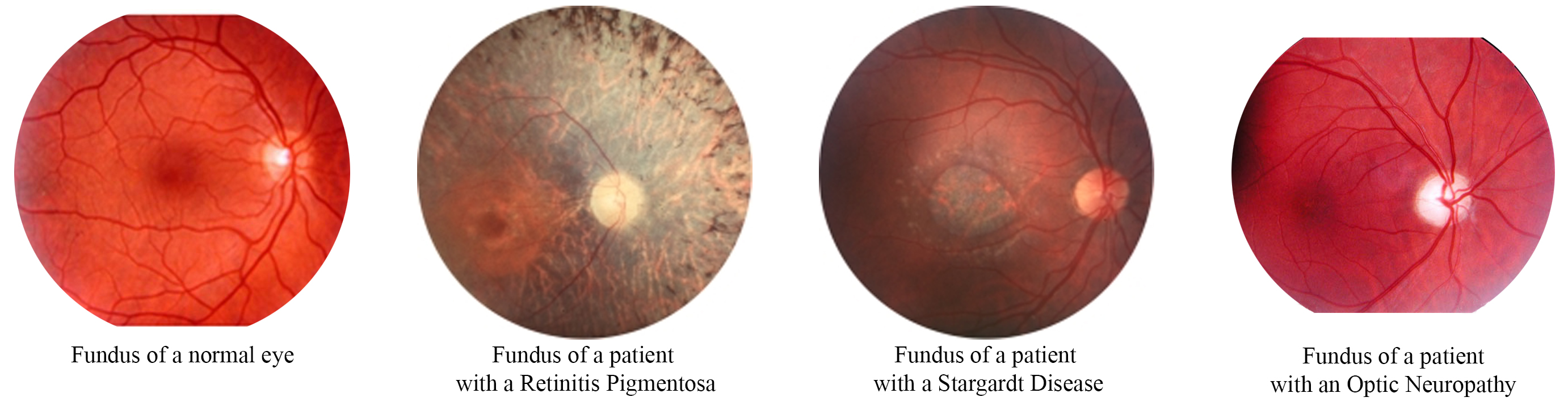
Currently, we are screening large cohorts of patients with either autosomal dominant or autosomal dominant inherited RP or hereditary optic neuropathies. Several new potential genes are under study.
-
Role of the interphotoreceptor matrix in the vitelliform macular dystrophies and RP
The retina, located at the posterior part of the eye, is divided into several layers with specific functions. In particular, photoreceptors (PR) convert light into an electrical signal and retinal pigment epithelium (RPE) that plays a role in the maintenance and survival of PR. The interactions between RPE and PR are mainly at the level of the interphotoreceptor matrix (IPM), a matrix that allows the junction between the outer segments of the PR and the apical micovilli of the RPE. IPM is the place of nutrient exchange between RPE and PR and retinoid transit of the visual cycle.
In 2010, mutations in the IMPG2 gene (Interphotoreceptor Matrix ProteoGlycan 2) encoding SPACRCAN were identified in patients with autosomal recessive RP. In 2013, we showed that the IMPG1 gene (Interphotoreceptor Matrix ProteoGlycan 1) encoding SPACR is responsible for dominant and recessive forms of vitelliform macular dystrophy (VMD, Manes et al., 2013). Our group has also identified mutations in IMPG2 in rare cases of VMD (Meunier et al., 2014). The involvement of SPACR and SPACRCAN in RP and VMD shows the major role of IPM in retinal pathophysiology.
The project aims to determine how a defect or absence of SPACR and SPACRCAN proteins modify the properties of IPM to lead to lipofuscin accumulation and PR death.
Major Publications
Olivier G, et al., J Med Genet, 2020-107150, 2020
Manes G, et al., Hum Mol Genet, 26:4367–4374, 2017
Manes G, et al., Mol Vis, 23:198-209, 2017
Meunier I, et al., Hum Mol Genet, 2016
Manes G., et al., Am J Ophthalmol, 159(2):302-14, 2015
Meunier I., et al., Ophthalmology, 121(12):2406-14, 2014
Manes G, et al., PLoS One 23;9(4), 2014
Manes G., et al., Am J Hum Genet, 93, 571-578, 2013
Bocquet B, et al., Mol Vis, 19:2487-2500, 2013
Hamel C., et al., Am J Ophthalmol, 147, 609-620, 2009
Delettre C., et al., Nat Genet, 26, 207-210, 2000
Collaborations
- National Reference Centrer Rare Diseases of Montpellier Maolya : I. Meunier, C. Blanchet, A. Roubertie)
- European Retinal Dystrophy Consortium: C. Ayuso (Spain), S. Banfi (Italy), F. Cremers (the Netherlands), C. Inglehearn (Great Britain), B. Leroy (Belgium), C. Rivolta (Switzerland), D. Sharon (Israel), B. Wissinger (Germany).
- French national reference and competence centers for retinal dystrophies: S. Defoort-Dhellemmes (Lille), H. Dollfus (Strasbourg), J. Kaplan (Paris), J. Sahel (Paris), M. Weber (Nantes), X. Zanlonghi (Nantes).
- Département de génétique médicale, maladies rares et médecine personnalisée, CHU de Montpellier : T. Guignard
Financements



