")
")










The retina is particularly amenable to gene therapy because it is accessible via relatively non-invasive routes, it is small and enclosed allowing the use of small vector doses, and it is immuno-privileged due to sequestration from the systemic circulation by the blood-retina barrier. Moreover, inherited retinal dystrophies (IRDs) are favourable candidates for gene therapy because they are often monogenic, have characteristic clinical signs allowing an early diagnosis, and progress slowly to blindness allowing a large therapeutic window.
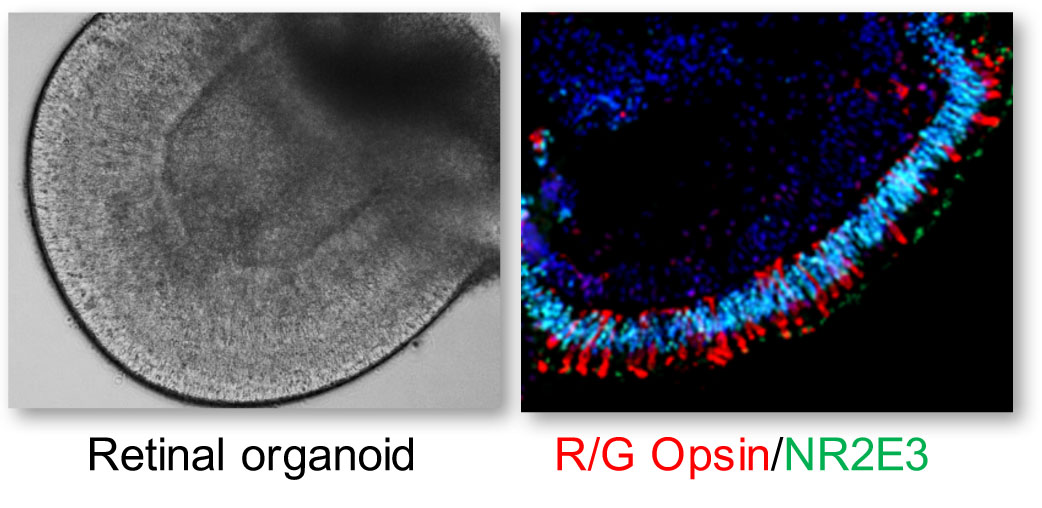
The aim of our work is to generate and use human retinal cell models of IRDs to confirm causative variants, study pathophysiology and develop innovative therapies. To generate such models, we take skin fibroblasts from a patient carrying mutations in a gene of interest and reprogram these cells into induced pluripotent stem cells (iPSC). These disease-specific iPSC are then differentiated into retinal pigment epithelium (RPE) or retinal organoids containing photoreceptors.
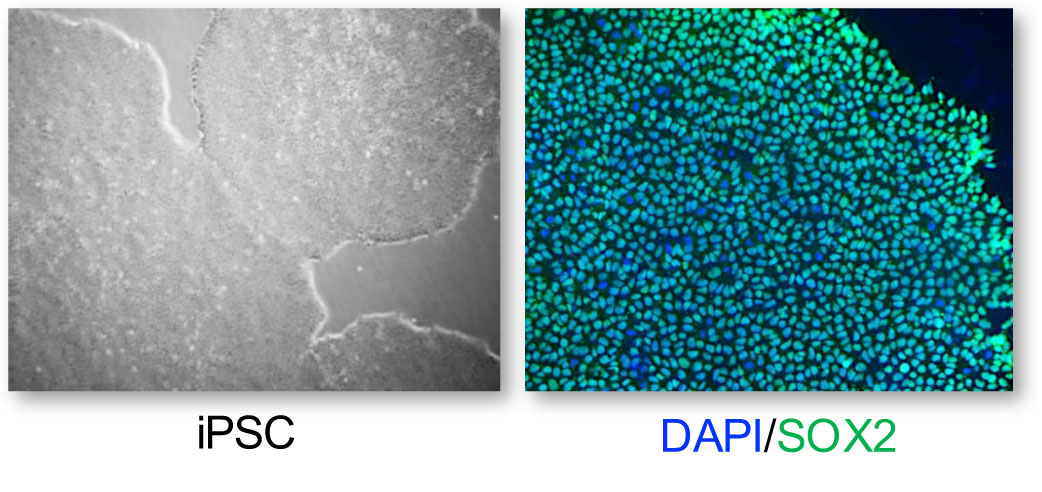
Our models are focused on a variety of IRDs: choroideremia due to mutations in the CHM gene, retinitis punctata albescens due to RLBP1, Usher syndrome type 2 due to USH2A, autosomal dominant retinitis pigmentosa due to NR2E3, as well as the various clinical forms associated with either CRX or ABCA4. In addition to allowing a comprehensive genotype-phenotype correlation, these models provide the basis of our proof-of-concept therapeutic studies, which comprise gene supplementation approaches as well as allele correction or invalidation by CRISPR/Cas9 genome editing.

Major publications
Sanjurjo-Soriano C et al, Mol Ther Methods Clin Dev. 17:156-173, 2019
Erkilic N et al, Cells 8:1068, 2019
Torriano S et al, Hum Mol Genet. 26:3573-3584, 2017
Cereso N et al, Mol Ther Methods Clin Dev. 1: 14011, 2014
Hippert C et al, Mol Ther. 16: 1372-1381, 2008
Kalatzis V et al, Hum Mol Genet. 13: 1361-1371, 2004
Kalatzis V et al, EMBO J. 20:5940-5949, 2001
Kalatzis V et al, Nature Genet. 15: 157-164, 1997
Collaborations
- Pr Ian MacDonald, Alberta University, Edmonton, Canada
- Dr. Yvan Arsenijevic, University of Lausanne, Switzerland
- Pr Carmen Ayuso, Fundacion Jimenez Diaz, Madrid, Spain
- Dr. Mariya Moosajee, University College London, UK
- Dr Anne-Françoise Roux, CHRU Montpellier, France
- Pr. Franck Pellestor, CHRU, Montpellier, France
- Pr. John De Vos, IRB, Montpellier, France
- Dr Sara Salinas, Inserm, Montpellier, France
Fundings

![]()

![]()
![]()
![]()









![]()








